1.
http://eurogenes.blogspot.ca/2012/01/eurogenes-north-euro-clusters-phase-2.html
2.
http://www.harappadna.org/category/chromopainter-2/
3.
http://blogs.discovermagazine.com/gnxp/2012/01/population-structure-using-haplotype-data/#.UYAD1HE-sRN
2013年4月30日星期二
Rscript example - using R to executable functions
I use Rscript as my prime scripting language to perform some command-line tasks. The reason for this is I don’t know much about bash scripts nor perl language. Writing a script in R is very simple if you know R language. But there are a few things need to consider.
- How to pass arguments to the script
- Which version of Rscript should you use
- A facility to provide a bash-line help
I did a simple try and here is an example Rscript how it works
Stamen maps with spplot
Stamen maps with spplot
Several R packages provide an interface to query map services (Google Maps,Stamen Maps or OpenStreetMap) to obtain raster images from them. As far as I know, there are three packages devoted to this task: RgoogleMaps,OpenStreetMap and ggmap. The latter two are increasingly popular with a wide collection of providers.

2013年4月26日星期五
yandell lab - on human population genetics
http://www.yandell-lab.org/software/index.html
 VAAST
VAAST
VAAST (the Variant Annotation, Analysis & Search Tool) is a probabilistic search tool for identifying damaged genes and their disease-causing variants in personal genome sequences. VAAST builds upon existing amino acid substitution (AAS) and aggregative approaches to variant prioritization, combining elements of both into a single unified likelihood-framework that allows users to identify damaged genes and deleterious variants with greater accuracy, and in an easy-to-use fashion. VAAST can score both coding and non-coding variants, evaluating the cumulative impact of both types of variants simultaneously. VAAST can identify rare variants causing rare genetic diseases, and it can also use both rare and common variants to identify genes responsible for common diseases. VAAST thus has a much greater scope of use than any existing methodology.
 MAKER 2 (updated 07-22-2012)
MAKER 2 (updated 07-22-2012)
MAKER is a portable and easily configurable genome annotation pipeline. It's purpose is to allow smaller eukaryotic and prokaryotic genomeprojects to independently annotate their genomes and to create genome databases. MAKER identifies repeats, aligns ESTs and proteins to a genome, produces ab-initio gene predictions and automatically synthesizes these data into gene annotations having evidence-based quality values. MAKER is also easily trainable: outputs of preliminary runs can be used to automatically retrain its gene prediction algorithm, producing higher quality gene-models on seusequent runs. MAKER's inputs are minimal and its ouputs can be directly loaded into a GMOD database. They can also be viewed in the Apollo genome browser; this feature of MAKER provides an easy means to annotate, view and edit individual contigs and BACs without the overhead of a database. MAKER should prove especially useful for emerging model organism projects with minimal bioinformatics expertise and computer resources.
 RepeatRunner
RepeatRunner
RepeatRunner is a CGL-based program that integrates RepeatMasker with BLASTX to provide a comprehensive means of identifying repetitive elements. Because RepeatMasker identifies repeats by means of similarity to a nucleotide library of known repeats, it often fails to identify highly divergent repeats and divergent portions of repeats, especially near repeat edges. To remedy this problem, RepeatRunner uses BLASTX to search a database of repeat encoded proteins (reverse transcriptases, gag, env, etc...). Because protein homologies can be detected across larger phylogenetic distances than nucleotide similarities, this BLASTX search allows RepeatRunner to identify divergent protein coding portions of retro-elements and retro-viruses not detected by RepeatMasker. RepeatRunner merges its BLASTX and RepeatMasker results to produce a single, comprehensive XML-based output. It also masks the input sequence appropriately. In practice RepeatRunner has been shown to greatly improve the efficacy of repeat identifcation. RepeatRunner can also be used in conjunction with PILER-DF - a program designed to identify novel repeats - and RepeatMasker to produce a comprehensive system for repeat identification, characterization, and masking in the newly sequenced genomes.
ImagePlane
ImagePlane is python based image analysis software designed for the automated analysis of images of the animal S. mediterranea. This software allows the animals's neoblasts to be quantified and tested for assymetries along its veritcal and hoizontal axes. ImagePlane also allows simple mophology categorizations to be made based on the overall shape of the animal.
 CGL
CGL
CGL is a software library designed to facilitate the use of genome annotations as substrates for computation and experimentation; we call it "CGL", an acronym for Comparitive Genomics Library, and pronounce it "Seagull". The purpose of CGL is to provide an informatics infrastructure for a laboratory, department, or research institute engaged in the large-scale analysis of genomes and their annotations.
2013年4月25日星期四
GateOne - a web-based Terminal Emulator and SSH client
http://liftoffsoftware.com/Products/GateOne
a web-based Terminal Emulator and SSH client that brings the power of the command line to the web. It requires no browser plugins and is built on top of a powerful plugin system that allows every aspect of its appearance and functionality to be customized.
a web-based Terminal Emulator and SSH client that brings the power of the command line to the web. It requires no browser plugins and is built on top of a powerful plugin system that allows every aspect of its appearance and functionality to be customized.
duplicate/repeat a character times in terminal
# generate many times of "S"
for i in $(seq 303738); do echo -n 'S'; done; printf "\n" > multi_S
transpose a matrix (from columns to row)
# transpose a matrix (from columns to row)
awk '{for (i=1; i<=NF; i++) a[i]=a[i](NR!=1?FS:"")$i} END {for (i=1; i in a; i++) print a[i]}' file
insert contents of a file to another file at specific line
# insert content of "mao" to file "head", after the line with "source"
sed '/source/r mao' head | less -S
2013年4月24日星期三
DNAPlotter: circular and linear interactive genome visualization
http://bioinformatics.oxfordjournals.org/content/25/1/119.full

Circular and linear DNA diagrams provide a powerful tool for illustrating the features of a genome in their full genomic context. CGView (Stothard and Wishart,2005) is a circular genome viewer that produces PNG, JPEG or scalable vector graphics (SVG) format. It is primarily designed to be part of a pipeline and not an interactive and editable viewer. GenomeDiagram (Pritchard et al., 2006) is a Python module, which can also be used to generate diagrams. GenomePlot (Gibson and Smith, 2003), GenoMap (Sato and Ehira, 2003) and Microbial Genome Viewer (MGV; Kerkhoven et al., 2004), are GUI based but do not provide the ability to directly read in files in the common sequence formats or a filtering tool to define tracks.
Circos (http://mkweb.bcgsc.ca/circos/) is web-based genome comparison visualizer that is configured from flat files. It can be configured for comparisons within a genome or between multiple genomes. There are also commercial packages available that draw circular and linear plots.
Below we present DNAPlotter, a collaborative project combining graphics from the Jemboss (http://emboss.sf.net/Jemboss/) DNA viewer and Artemis (http://www.sanger.ac.uk/Software/Artemis/) libraries to read files and filter features, which has been designed to be a graphical means of customizing circular and linear diagrams.
2013年4月23日星期二
Crossing the species barrier: genomic hotspots of introgression between two highly divergent Ciona intestinalis species
http://mbe.oxfordjournals.org/content/early/2013/04/05/molbev.mst066.full.pdf+html
ABC running with sliding-windows
Inferring a realistic demographic model from genetic data is an important challenge to gain insights into the historical events during the speciation process and to detect molecular signatures of selection along genomes. Recent advances in divergence population genetics have reported that speciation in face of gene flow occurred more frequently than theoretically expected, but the approaches used did not account for genome wide heterogeneity (GWH) in introgression rates. Here we investigate the impact of GWH on the inference of divergence with gene flow between two cryptic species of the marine modelCiona intestinalis by analysing polymorphism and divergence patterns in 852 protein-coding sequence loci.
These morphologically similar entities are highly diverged molecular-wise, but evidence of hybridisation has been reported in both laboratory and field studies. We compare various speciation models and test for GWH under the ABC framework. Our results demonstrate the presence of significant extents of gene flow resulting from a recent secondary contact after >3 My of divergence in isolation. The inferred rates of introgression are relatively low, highly variable across loci and mostly unidirectional, which is consistent with the idea that numerous genetic incompatibilities have accumulated over time throughout the genomes of these highly-diverged species. A genomic map of the level of gene flow identified two hotspots of introgression, i.e. large genome regions of unidirectional introgression. This study clarifies the history and degree of isolation of two cryptic and partially sympatric model species, and provides a methodological framework to investigate GWH at various stages of speciation process.
CodonPhyML: Fast Maximum Likelihood Phylogeny Estimation under Codon Substitution Models
http://mbe.oxfordjournals.org/content/early/2013/04/02/molbev.mst034.abstract
Markov models of codon substitution naturally incorporate the structure of the genetic code and the selection intensity at the protein level, providing a more realistic representation of protein-coding sequences compared with nucleotide or amino acid models. Thus, for protein-coding genes, phylogenetic inference is expected to be more accurate under codon models. So far, phylogeny reconstruction under codon models has been elusive due to computational difficulties of dealing with high dimension matrices. Here, we present a fast maximum likelihood (ML) package for phylogenetic inference, CodonPhyML offering hundreds of different codon models, the largest variety to date, for phylogeny inference by ML. CodonPhyML is tested on simulated and real data and is shown to offer excellent speed and convergence properties. In addition, CodonPhyML includes most recent fast methods for estimating phylogenetic branch supports and provides an integral framework for models selection, including amino acid and DNA models.
Markov models of codon substitution naturally incorporate the structure of the genetic code and the selection intensity at the protein level, providing a more realistic representation of protein-coding sequences compared with nucleotide or amino acid models. Thus, for protein-coding genes, phylogenetic inference is expected to be more accurate under codon models. So far, phylogeny reconstruction under codon models has been elusive due to computational difficulties of dealing with high dimension matrices. Here, we present a fast maximum likelihood (ML) package for phylogenetic inference, CodonPhyML offering hundreds of different codon models, the largest variety to date, for phylogeny inference by ML. CodonPhyML is tested on simulated and real data and is shown to offer excellent speed and convergence properties. In addition, CodonPhyML includes most recent fast methods for estimating phylogenetic branch supports and provides an integral framework for models selection, including amino acid and DNA models.

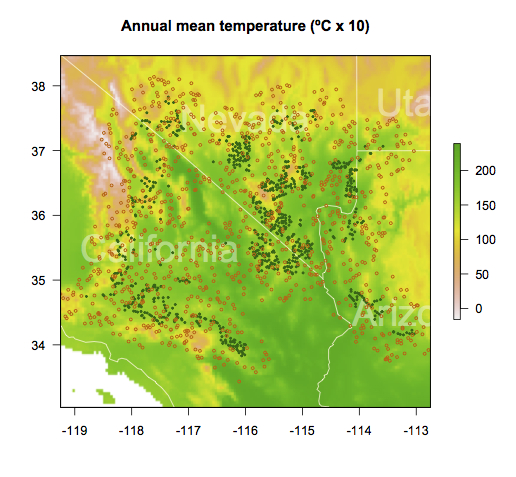

2013年4月22日星期一
Interspecific Divergence of Transcription Networks along Lines of Genetic Variance in Drosophila: Dimensionality, Evolvability and Constraint
Interspecific Divergence of Transcription Networks along Lines of Genetic Variance inDrosophila: Dimensionality, Evolvability and Constraint
Change in gene expression is a major facilitator of phenotypic evolution. Understanding the evolutionary potential of gene expression requires taking into account complex systems of regulatory networks, the structure of which could potentially bias evolutionary trajectories. We analyzed the evolutionary potential and divergence of multigene expression in three well-characterized signaling pathways in Drosophila, the mitogen-activated protein kinase (MapK), the Toll, and the insulin receptor/Foxo (InR/Foxo or InR/TOR) pathways in a multivariate quantitative genetic framework. Gene expression data from a natural population of D. melanogaster were used to estimate the genetic variance–covariance matrices (G) for each network. Although most genes within each pathway exhibited significant genetic variance, the number of independent dimensions of multivariate genetic variance was fewer than the number of genes analyzed. However, for expression, the reduction in dimensionality was not as large as seen for other trait types such as morphology. We then tested whether gene expression divergence between D. melanogaster and an additional six species of the Drosophila genus was biased along the major axes of standing variation observed in D. melanogaster. In many cases, divergence was restricted to directions of phenotypic space harboring above average levels of genetic variance in D. melanogaster, indicating that genetic covariances between genes within pathways have biased interspecific divergence. We tested whether co-expression of genes in both sexes has also biased the pattern of divergence. Including cross-sex genetic covariances increased the degree to which divergence was biased along major axes of genetic variance, suggesting that the co-expression of genes in males and females can generate further constraints on divergence across the Drosophila phylogeny. In contrast to patterns seen for morphological traits in vertebrates, transcriptional constraints do not appear to break down as divergence time between species increases, instead they persist over tens of millions of years of divergence.
2013年4月21日星期日
OpenStreetMap package of R - get beautiful maps
The bigest change in the new version is the addition of dozens of tile servers, giving the user the option of many different map looks, including those from Bing, MapQuest and Apple.
nm = c("osm", "maptoolkit-topo",
"waze", "mapquest", "mapquest-aerial",
"bing", "stamen-toner", "stamen-terrain",
"stamen-watercolor", "osm-german", "osm-wanderreitkarte",
"mapbox", "esri", "esri-topo",
"nps", "apple-iphoto", "skobbler",
"opencyclemap", "osm-transport",
"osm-public-transport", "osm-bbike", "osm-bbike-german")
png(width=1200,height=2200)
par(mfrow=c(6,4))
for(i in 1:length(nm)){
print(nm[i])
map = openmap(c(lat= 38.05025395161289, lon= -123.03314208984375),
c(lat= 36.36822190085111, lon= -120.69580078125),
minNumTiles=9,type=nm[i])
plot(map)
title(nm[i],cex.main=4)
}
dev.off()

a Little Book of R for Bioinformatics
http://a-little-book-of-r-for-bioinformatics.readthedocs.org/en/latest/#
Chapters in this Book
- How to install R and a Brief Introduction to R
- Neglected Tropical diseases
- DNA Sequence Statistics (1)
- DNA Sequence Statistics (2)
- Sequence Databases
- REVISION EXERCISES 1
- Pairwise Sequence Alignment
- Multiple Alignment and Phylogenetic trees
- REVISION EXERCISES 2
- Computational Gene-finding
- Comparative Genomics
- Hidden Markov Models
- Answers to the End-of-chapter Exercises
- Answers to Revision Exercises
- Protein-Protein Interaction Graphs
R 3.0.0 - a new R edition
1.
http://nsaunders.wordpress.com/2013/04/04/a-brief-note-r-3-0-0-and-bioinformatics/
2. install R, in windows and linux and new features of this new R
http://www.r-statistics.com/
http://nsaunders.wordpress.com/2013/04/04/a-brief-note-r-3-0-0-and-bioinformatics/
2. install R, in windows and linux and new features of this new R
http://www.r-statistics.com/
2013年4月17日星期三
A wide extent of inter-strain diversity in virulent and vaccine strains of alpha-herpesviruses
Good example on de novo genome assembly, annotation, comparsion
http://genomics-pubs.princeton.edu/prv/scripts.shtml
http://genomics-pubs.princeton.edu/prv/scripts.shtml
Overview & Table of Contents
Part I - Pre-Assembly Processing
These steps take raw Illumina® sequence data and prepare it for de novo assembly, using a series of quality-controls and filters.
Part II - De novo Assembly & Genome Finishing
This phase of genome production takes high-quality short sequence reads, and by homology-based overlaps, joins them into longer and longer pieces.
Part III - Quality Assessment of New Genomes
These steps assess the quality of the assembly and address any inconsistencies. These scripts function in conjunction with wet-bench analyses, such as restriction fragment length polymorphism (RFLP) analysis and directed PCR validations.
Part IV - Annotation & Genome Comparison
This phase involves annotation, curation, and comparison of multiple genome sequences to reveal interesting biological features.
Addendum - Relevant Websites and Lists of Scripts Used
All scripts used are freely available online (web links provided) or are available here (see Downloads link on left) for download and free use.
2013年4月16日星期二


Stothard Research Group
http://www.ualberta.ca/~stothard/software.html
align_learn.pl - this Perl script converts a multiple sequence alignment into a format that can be readily analyzed using common machine learning algorithms. Specifically, the program accepts a sequence alignment in FASTA format and converts it into an ARFF (attribute-relation file format) file containing data attributes and data instances. The ARFF format can then be read by the Weka machine learning software, which provides implementations of many machine learning algorithms.
Developed by: Paul Stothard.
Availability: align_learn.zip, align_learn.tar.gz.
Developed by: Paul Stothard.
Availability: align_learn.zip, align_learn.tar.gz.
annotate_SNPs.pl - this Perl script annotates SNPs identified by the next-generation sequencing of genomic DNA or transcripts. It is designed to accept SNPs from the AB diBayes SNP package, Maq, or any other program that can provide a reference sequence identifier, a SNP position, the base found at that position in the reference sequence, and the bases found at that position among the sequencing reads. The script examines each SNP record provided as input and uses Ensembl, NCBI, and EBI to provide a detailed description of its expected functional significance, regardless of whether or not the SNP has been previously described. Included in this description is a functional class ('NON_SYNONYMOUS_CODING' for example), and information about the affected transcript, if applicable, including transcript ID, position of the SNP, the alleles as they would appear in the transcript sequence, and Gene Ontology information. For protein-altering SNPs, each of the resulting protein sequences is compared to orthologous proteins, to determine which of the alleles most drastically changes the protein. Using a locally installed version of Ensembl this script can annotate 4,000,000 SNPs in about two days on a standard desktop system. Thus this script is suitable for annotating the SNPs arising from genome resequencing projects.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
backup.sh - this shell script archives directories of interest on a Linux-based system. When it is first run, and on the first of each month, this script generates a full backup of the files and directories listed in the include.conf file. Files and directories listed in the exclude.conf file are not included in the archive. These full backups are not overwritten by future backups. Each Sunday the script performs a full backup that is overwritten the following Sunday. Every day the script performs an incremental backup, storing the files that have changed since the last full backup. These incremental backup files are named after the day of the week they are performed, and are overwritten each week. The script sends an email on the first of each month, and whenever any backup fails. The script splits each full backup into a series of smaller files, suitable for burning to CD or DVD. When a full backup is generated, the MD5 hash value of the complete backup file is written to a README file in the same directory as the backup files. Included in the README are directions for assembling the split backup files into the original file.
Developed by: Paul Stothard.
Availability: backup_script.zip, backup_script.tar.gz.
Developed by: Paul Stothard.
Availability: backup_script.zip, backup_script.tar.gz.
blast_hit_features.pl - this Perl script accepts BLAST results obtained from local_blast_client.pl or remote_blast_client.pl. The results must have been obtained using blastn, tblastn, or tblastx searches (i.e. nucleotide databases), since this script uses GenBank files to obtain feature information for sequence hits. For each entry in the BLAST results, the GI number of the hit, if available, is used to obtain the corresponding sequence record from NCBI, in GenBank format. The features in the GenBank file are compared to the coordinates of the HSP, and features overlapping with the HSP are added to the existing BLAST results. The modified results are written to a new file. The three nearest features preceding the HSP (located to the left of the HSP) and the three nearest features located after the HSP (located to the right of the HSP) are also added to the output.
Developed by: Paul Stothard.
Availability: blast_hit_features.zip, blast_hit_features.tar.gz.
Developed by: Paul Stothard.
Availability: blast_hit_features.zip, blast_hit_features.tar.gz.
blast_hit_flanking_sequence.pl - this Perl script accepts blastn search results obtained from local_blast_client.pl or remote_blast_client.pl. In addition to the BLAST results file, the script requires the query sequences and database sequences in FASTA format. For each BLAST result, the script constructs a modified query sequence, in which the query is extended using sequence extracted from the hit sequence. The amount of hit sequence added to the ends of the query can be specified using the -u and -d options.
Developed by: Paul Stothard.
Availability: blast_hit_flanking_sequence.zip, blast_hit_flanking_sequence.tar.gz.
Developed by: Paul Stothard.
Availability: blast_hit_flanking_sequence.zip, blast_hit_flanking_sequence.tar.gz.
blast_hits_in_ucsc_genome_browser.pl - this Perl script accepts BLAST results obtained from local_blast_client.pl or remote_blast_client.pl. The results must have been obtained using blastn, tblastn, or tblastx searches (i.e. nucleotide databases) and database sequences downloaded from the UCSC Genome Browser site (http://hgdownload.cse.ucsc.edu/downloads.html). The BLAST results are converted to annotation files for the UCSC Genome Browser, and a separate HTML file containing links to each feature in the annotation files is created. Clicking on a link in the HTML file loads the genome region involving the BLAST HSP into the UCSC Genome Browser and passes the annotations in the relevant annotation file to the browser for inclusion in the view.
Developed by: Paul Stothard.
Availability: blast_hits_in_ucsc_genome_browser.zip, blast_hits_in_ucsc_genome_browser.tar.gz.
Developed by: Paul Stothard.
Availability: blast_hits_in_ucsc_genome_browser.zip, blast_hits_in_ucsc_genome_browser.tar.gz.
build_cluster_script.pl - this Perl script creates an executable shell script with the specified command repeated n number of times. Every occurrence of the '$' symbol in the command is replaced by a number, from 1 to n. Alternatively, the "-l" option causes letters to be used in place of numbers (eg 'aa' instead of '1', 'ab' instead of '2'). This script can be used to generate scripts for batch processing on a computer cluster.
Developed by: Jason Grant and Paul Stothard.
Availability: build_cluster_script.zip, build_cluster_script.tar.gz.
Developed by: Jason Grant and Paul Stothard.
Availability: build_cluster_script.zip, build_cluster_script.tar.gz.
cDNA_library_entropy.pl - this Perl script accepts a directory containing one or more sequence files in multi-FASTA format. Typically each file will contain the sequences obtained from a single EST library or tissue type. By default the script looks for a UniGene annotation identifier in each sequence title, for example 'Bt.22094'. A different ID type can be specified using the -m option. The number of sequences present for each ID is determined. The script uses these counts to calculate the information entropy of the library in bits. This value increases as the number of distinct sequences in a library increases, and decreases as the number of replicates of a particular sequence increases. The -d option can be used to obtain the information entropy of combinations of libraries. For example, specifying '-d 2' causes all possible combinations of two libraries to be evaluated. This script is intended to aid in the selection of tissues for SNP discovery by mRNA sequencing.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
CGView - a Java package for generating high quality, zoomable maps of circular genomes. Its primary purpose is to serve as a component of sequence annotation pipelines. Feature information and rendering options are supplied to the program using an XML file, a tab delimited file, or an NCBI ptt file. CGView converts the input into a graphical map (PNG, JPG, or Scalable Vector Graphics format), complete with labels, a title, and legends. In addition to the default full view map, the program can generate a series of hyperlinked maps showing expanded views. The linked maps can be explored using any web browser, allowing rapid genome browsing, and facilitating data sharing. The feature labels in maps can be hyperlinked to external resources, allowing CGView maps to be integrated with existing web site content or databases.
Developed by: Paul Stothard.
Availability: http://bioinformatics.org/cgview/
Developed by: Paul Stothard.
Availability: http://bioinformatics.org/cgview/
CGView Comparison Tool (CCT) - a package for visually comparing bacterial, plasmid, chloroplast, or mitochondrial sequences of interest to existing genomes or sequence collections. The comparisons are conducted using BLAST, and the BLAST results are presented in the form of graphical maps that can also show sequence features, gene and protein names, COG category assignments, and sequence composition characteristics. CCT can generate maps in a variety of sizes, including 400 Megapixel maps suitable for posters. Comparisons can be conducted within a particular species or genus, or all available genomes can be used. The entire map creation process, from downloading sequences to redrawing zoomed maps, can be completed easily using scripts included with the CCT. User-defined features or analysis results can be included on maps, and maps can be extensively customized. To simplify program setup, a CCT virtual machine that includes all dependencies preinstalled is available. Detailed tutorials illustrating the use of CCT are included with the CCT documentation.
Developed by: Paul Stothard and Jason Grant.
Availability: http://stothard.afns.ualberta.ca/downloads/CCT/
Developed by: Paul Stothard and Jason Grant.
Availability: http://stothard.afns.ualberta.ca/downloads/CCT/
cgview_xml_builder.pl - this Perl script accepts a variety of input files pertaining to circular genomes and generates an XML file for the CGView genome drawing program. This script can create the XML to display a variety of sequence composition plots, gene expression data, COG information, BLAST results, and more. See the included README file for additional information.
Developed by: Paul Stothard.
Availability: cgview_xml_builder.zip, cgview_xml_builder.tar.gz.
Developed by: Paul Stothard.
Availability: cgview_xml_builder.zip, cgview_xml_builder.tar.gz.
combine_output_files.pl - this Perl script combines files that are part of a file series (created by split_records.pl for example). Several options are avialable for controlling how comments and header lines are handled. This script can be used to combine results files generated on a computer cluster.
Developed by: Paul Stothard.
Availability: combine_output_files.zip, combine_output_files.tar.gz.
Developed by: Paul Stothard.
Availability: combine_output_files.zip, combine_output_files.tar.gz.
genome_pattern_search.pl - a Perl program that reads a genomic sequence in FASTA format and searches for the patterns you specify using regular expressions. A summary is generated for each sequence match, including: the sequence fragment that matched the pattern; the position of the first base; the position of the last base; the strand on which the match was found; the name of the gene containing the match or "not in gene"; the name of the nearest downstream gene; a description of the gene; the distance to the nearest downstream gene; the total times this exact sequence was found; the percentage of the instances of this exact sequence that were found inside of genes; and the average number of base pairs to the downstream gene for this exact sequence.
Developed by: Paul Stothard.
Availability: genome_pattern_search.zip, genome_pattern_search.tar.gz.
Developed by: Paul Stothard.
Availability: genome_pattern_search.zip, genome_pattern_search.tar.gz.
get_cds.pl - this Perl script accepts a GenBank or EMBL file and extracts the protein translations or the DNA coding sequences and writes them to a new file in FASTA format. Information indicating the reading frame and position of the coding sequence relative to the source sequence is added to the titles.
Developed by: Paul Stothard.
Availability: get_cds.zip, get_cds.tar.gz.
Developed by: Paul Stothard.
Availability: get_cds.zip, get_cds.tar.gz.
get_genes_in_area.pl - this Perl script accepts as input a position or list of positions in a genome and returns descriptions of nearby genes. The descriptions include position and function information, along with identifiers that can be used to access related records in other databases.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
get_orfs.pl - this Perl script accepts a sequence file as input and extracts the open reading frames (ORFs) greater than or equal to the size you specify. The resulting ORFs can be returned as DNA sequences or as protein sequences. The titles of the sequences include start, stop, strand, and reading frame information. The sequence numbering includes the stop codon (when encountered) but the translations do not include a stop codon character.
Developed by: Paul Stothard.
Availability: get_orfs.zip, get_orfs.tar.gz.
Developed by: Paul Stothard.
Availability: get_orfs.zip, get_orfs.tar.gz.
get_snps_by_gene_ontology.pl - this Perl script accepts a species name and a Gene Ontology (GO) accession number, and returns a list of SNPs located in or nearby genes associated with the GO accession. Several fields of information are provided for each SNP, including ID, location, flanking sequence, and alleles. Gene and transcript identifiers and descriptions of gene function are also provided.
Developed by: Paul Stothard.
Availability: get_snps_by_gene_ontology.zip, get_snps_by_gene_ontology.tar.gz.
Developed by: Paul Stothard.
Availability: get_snps_by_gene_ontology.zip, get_snps_by_gene_ontology.tar.gz.
local_blast_client.pl - this Perl script accepts a FASTA file containing multiple sequences as input. It then submits each sequence to a locally installed version of the blastall program. For each of the hits obtained, the script retrieves a descriptive title by performing a separate Entrez search of NCBI's databases. Each BLAST hit and its descriptive title are written to a single tab-delimited output file.
Developed by: Paul Stothard.
Availability: local_blast_client.zip, local_blast_client.tar.gz.
Developed by: Paul Stothard.
Availability: local_blast_client.zip, local_blast_client.tar.gz.
maq_pipeline.sh - this bash script processes short sequence reads from Illumina's Genome Analyzer (Solexa) system, using the Maq package. The script automates the entire analysis process, and parallelizes the most intensive analysis step if run on a computing cluster with Sun Grid Engine.
Developed by: Paul Stothard.
Availability: maq_pipeline.zip, maq_pipeline.tar.gz.
Developed by: Paul Stothard.
Availability: maq_pipeline.zip, maq_pipeline.tar.gz.
md5_sums.pl - this Perl script accepts a list of directories and recursively generates a list of the files in the directories and their MD5 values. An optional list of directories and files to exclude from the calculation can also be supplied. The MD5 calculation can be skipped for large files, using the optional size parameter.
Developed by: Jason Grant.
Availability: md5_sums.zip, md5_sums.tar.gz.
Developed by: Jason Grant.
Availability: md5_sums.zip, md5_sums.tar.gz.
ncbi_monitor.pl - this Perl script performs NCBI Entrez searches to identify publications related to genomic regions of interest in a species of interest. More specifically, this script accepts an organism name, chromosome name, and base position as input. It then retrieves the IDs for all Entrez Gene records located within a certain distance of the base position (the distance can be adjusted using the -f option). For each Gene record the script obtains IDs of PubMed records identified by NCBI as being related to the Gene record. If the script has previously written output to the specified output directory (i.e. the directory supplied using the -o option), it examines the previously obtained PubMed IDs to see which IDs are new. An email message describing the newly obtained records is then sent to the email address supplied using the -e option. The PubMed results are also written to a file in the output directory. If the -h option is specified, NCBI's HomoloGene database is also queried for each Gene record, in an attempt to obtain additional PubMed records, linked to the HomoloGene hits. These PubMed records may describe results obtained in other species, but could be relevant nonetheless.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
ncbi_search.pl - this Perl script uses NCBI's Entrez Programming Utilities to perform searches of NCBI databases. The script can return complete database records, or sequence IDs.
Developed by: Paul Stothard.
Availability: ncbi_search.zip, ncbi_search.tar.gz.
Developed by: Paul Stothard.
Availability: ncbi_search.zip, ncbi_search.tar.gz.
NGS-SNP - this collection of scripts annotates raw SNP lists returned from programs such as Maq. SNPs are classified as synonymous, nonsynonymous, 3' UTR, etc. regardless of whether or not they match existing SNP records. Included among the annotations, several of which are not available from any existing SNP annotation tools, are the results of detailed comparisons with orthologous sequences. These comparisons allow, for example, SNPs to be sorted or filtered based on how drastically the SNP changes the score of a protein alignment. Other fields indicate whether or not the SNP-altered residue exhibits co-evolution with other residues in the protein, the names of overlapping protein domains or features, and the conservation of both the SNP site and flanking regions. NCBI, Ensembl, and Uniprot IDs are provided for genes, transcripts, and proteins when applicable, along with Gene Ontology terms, a gene description, phenotypes linked to the gene, and an indication of whether the SNP is novel or known. A "Model_Annotations" field provides several annotations obtained by transferring in silico the SNP to an orthologous gene, typically in a well-characterized species.
Developed by: Paul Stothard, Jason Grant, and Xiaoping Liao.
Availability: NGS-SNP.
Developed by: Paul Stothard, Jason Grant, and Xiaoping Liao.
Availability: NGS-SNP.
obtain_reference_transcripts.pl - this Perl script builds a FASTA file consisting of the canonical transcripts for all the genes in Ensembl for a given organism. The canonical transcript is defined as either the longest CDS (if the gene encodes a protein), or as the longest cDNA. Ensembl gene entries can be associated with many transcripts--this script aims to get the "best" single transcript for each gene. The resulting FASTA file is suitable for sequence searches and for mapping sequence reads derived from cDNAs. The -a option can be used to specify that all transcripts should be downloaded, not just the canonical ones.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
obtain_reference_chromosomes.pl - this Perl script builds a FASTA file consisting of the chromosome sequences in Ensembl for a given organism. The resulting FASTA file is suitable for sequence searches and for mapping sequence reads.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
Developed by: Paul Stothard.
Availability: Included in the NGS-SNP package.
random_sequence_reads.pl - this Perl script generates simulated sequence reads from a file of sequences in FASTA format. The starting position of each read is chosen at random. The length of the reads is specified using the -L option. Reads truncated because the end of a sequence is encountered are discarded if they are shorter than the length specified using the -m option. Sampling is continued until the desired number of reads is obtained.
Developed by: Paul Stothard.
Availability: random_sequence_reads.zip, random_sequence_reads.tar.gz.
Developed by: Paul Stothard.
Availability: random_sequence_reads.zip, random_sequence_reads.tar.gz.
random_sequence_sample.pl - this Perl script selects a random sample of sequences from a FASTA file containing multiple sequences. The sample is written to a new text file. Sampling can be performed with or without replacement.
Developed by: Paul Stothard.
Availability: random_sequence_sample.zip, random_sequence_sample.tar.gz.
Developed by: Paul Stothard.
Availability: random_sequence_sample.zip, random_sequence_sample.tar.gz.
remote_blast_client.pl - this Perl script accepts a FASTA file containing multiple sequences as input. It submits each sequence to NCBI's BLAST servers, to identify related sequences in a database of interest. An optional 'limit by entrez query' value can be supplied to restrict the search. For each BLAST hit a descriptive title is obtained using a separate Entrez search. Each BLAST hit and its descriptive title are written to a single tab-delimited output file.
Developed by: Paul Stothard.
Availability: remote_blast_client.zip, remote_blast_client.tar.gz.
Developed by: Paul Stothard.
Availability: remote_blast_client.zip, remote_blast_client.tar.gz.
remote_in_silico_pcr.rb - this Ruby script accepts as input a list of primer sequences and uses the remote "UCSC In-Silico PCR" site to perform in silico PCR on the specified genome. By default only the top hit is returned for each primer pair--all the hits can be returned by using the '-m' option.
Developed by: Jason Grant.
Availability: remote_in_silico_pcr.zip, remote_in_silico_pcr.tar.gz.
Developed by: Jason Grant.
Availability: remote_in_silico_pcr.zip, remote_in_silico_pcr.tar.gz.
sequence_to_multi_fasta.pl - this Perl script reads a file consisting of a single DNA sequence (in raw, FASTA, GenBank, or EMBL format) and then divides the sequence into smaller sequences of the size you specify. The new sequences are written to a single output file with a modified title giving the position of the subsequence in relation to the original sequence. The new sequences are written in FASTA format.
Developed by: Paul Stothard.
Availability: sequence_to_multi_fasta.zip, sequence_to_multi_fasta.tar.gz.
Developed by: Paul Stothard.
Availability: sequence_to_multi_fasta.zip, sequence_to_multi_fasta.tar.gz.
space_check.sh - this shell script monitors hard drive space and sends an email when space becomes scarce. On the first day of each month the script sends an email report of hard drive space.
Developed by: Paul Stothard.
Availability: space_check.zip, space_check.tar.gz.
Developed by: Paul Stothard.
Availability: space_check.zip, space_check.tar.gz.
split_records.pl - this Perl script splits an input file into multiple output files, to allow analysis jobs to be divided among nodes in a computer cluster Several options are included for handling header lines, for specifying the record separator, and for controlling how files are named.
Developed by: Jason Grant and Paul Stothard.
Availability: split_records.zip, split_records.tar.gz.
Developed by: Jason Grant and Paul Stothard.
Availability: split_records.zip, split_records.tar.gz.
Sebas Ramos-Onsins lab
http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html
Analysis of Nucleotide Variability on Natural and Domesticated Populations:
Our focus is mainly on the species Sus scrofa (the pig) and close related species. We are interested in explaining the evolutionary processes involved in the history of this species and the effect of natural and artificial selection in wild and domestic breeds.
We are also interested in analyzing the variability in autopolyploid species using statistics adapted for pooled lineages.
Study and development of Neutrality tests and methods for statistical inference of evolutionary models:
A useful way to obtain a more interpretable information from raw sequencing data is the use of statistics and neutrality tests. They summarize the information observed and make possible to understand the evolutionary patterns more easily. We study and develop neutrality test to be used in evolutionary parameter inference.
Development of tools for the analysis of nucleotide variability:
Until now we have been working in developing tools for the analysis of nucleotide variability in multilocus data: analysis on multilocus data can be much precise in comparison to single locus because they reduce the variance of the parameters inferred in the analysis. We develop tools for multilocus population genetic analysis.
Now we are focusing on population genomics: massive parallel sequencing is revolutionizing the study of population genetics in many ways. We are developing bioinformatic tools for the analysis of variability at genomics level.
2013年4月8日星期一
Genome-Wide Survey of Pseudogenes in 80 Fully Re-sequenced Arabidopsis thaliana Accessions
Pseudogenes (Ψs), including processed and non-processed Ψs, are ubiquitous genetic elements derived from originally functional genes in all studied genomes within the three kingdoms of life. However, systematic surveys of non-processed Ψs utilizing genomic information from multiple samples within a species are still rare. Here a systematic comparative analysis was conducted of Ψs within 80 fully re-sequenced Arabidopsis thalianaaccessions, and 7546 genes, representing ~28% of the genomic annotated open reading frames (ORFs), were found with disruptive mutations in at least one accession. The distribution of these Ψs on chromosomes showed a significantly negative correlation betweenΨs/ORFs and their local gene densities, suggesting a higher proportion of Ψs in gene desert regions, e.g. near centromeres. On the other hand, compared with the non-Ψ loci, even the intact coding sequences (CDSs) in the Ψ loci were found to have shorter CDS length, fewer exon number and lower GC content. In addition, a significant functional bias against the null hypothesis was detected in the Ψs mainly involved in responses to environmental stimuli and biotic stress as reported, suggesting that they are likely important for adaptive evolution to rapidly changing environments by pseudogenization to accumulate successive mutations.
Microindel detection in short-read sequence data - comparing BWA with other aligners
The sensitivity of indel detection based on Novoalign and especially BWA alignments benefits from higher sequencing depth and read length.
http://bioinformatics.oxfordjournals.org/content/26/6/722.full
http://bioinformatics.oxfordjournals.org/content/26/6/722.full
2013年4月7日星期日
Eckertlab - on evolutionary genomics of trees
http://eckertlab.blogspot.ca/
the Eckert Lab located in the Department of Biology at Virginia Commonwealth University.
A plant evolutionary genomics lab.
Research topics range from the dissection of adaptive plant phenotypes into their genetic components to inferences of genome-wide patterns of polymorphism, divergence and natural selection.
http://evolfri.blogspot.ca/
the Eckert Lab located in the Department of Biology at Virginia Commonwealth University.
A plant evolutionary genomics lab.
Research topics range from the dissection of adaptive plant phenotypes into their genetic components to inferences of genome-wide patterns of polymorphism, divergence and natural selection.
http://evolfri.blogspot.ca/
2013年4月6日星期六
good GUI tool for sequences analysis
1. Unipro UGENE
http://ugene.unipro.ru/
http://bioinformatics.oxfordjournals.org/content/28/8/1166
http://ugene.unipro.ru/
http://bioinformatics.oxfordjournals.org/content/28/8/1166
- CAP3
- Bowtie-0.12.7
- BWA-0.5.9
- blast-2.2.25
- blast-2.2.25+
- clustalw-2.1
- mafft-6.847
- T-Coffee-8.99
- Mr.Bayes-3.2.0
Summary: Unipro UGENE is a multiplatform open-source software with the main goal of assisting molecular biologists without much expertise in bioinformatics to manage, analyze and visualize their data. UGENE integrates widely used bioinformatics tools within a common user interface. The toolkit supports multiple biological data formats and allows the retrieval of data from remote data sources. It provides visualization modules for biological objects such as annotated genome sequences, Next Generation Sequencing (NGS) assembly data, multiple sequence alignments, phylogenetic trees and 3D structures. Most of the integrated algorithms are tuned for maximum performance by the usage of multithreading and special processor instructions. UGENE includes a visual environment for creating reusable workflows that can be launched on local resources or in a High Performance Computing (HPC) environment. UGENE is written in C++ using the Qt framework. The built-in plugin system and structured UGENE API make it possible to extend the toolkit with new functionality.
Availability and implementation: UGENE binaries are freely available for MS Windows, Linux and Mac OS X at http://ugene.unipro.ru/download.html. UGENE code is licensed under the GPLv2; the information about the code licensing and copyright of integrated tools can be found in the LICENSE.3rd_party file provided with the source bundle.
订阅:
博文 (Atom)