The following content are scraped from this post.
#################################################
http://compbio.brc.iop.kcl.ac.uk/presentations/20100722-S.Newhouse.Next.Gen.Alignment.BRC.cluster.v1.pdf
Next Generation Sequence Alignment on the BRC Cluster
Illumina Paired-end , Alignment : BWA or Novoalign , SNP & Indel : GATK
Next Generation Sequence Alignment on the BRC Cluster
cmpfastq.pl : http://compbio.brc.iop.kcl.ac.uk/software/cmpfastq.php
Novoalign: http://www.novocraft.com
Genome Analysis Toolkit: http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
PICARD TOOLS: http://picard.sourceforge.net/
SAMTOOLS: http://samtools.sourceforge.net/
VCFTOOLS: http://vcftools.sourceforge.net/
FASTQ Files : http://en.wikipedia.org/wiki/FASTQ_format,
SAM/BAM Format : http://samtools.sourceforge.net/SAM1.pdf
PHRED Scores: http://en.wikipedia.org/wiki/Phred_quality_score
Next Generation Sequencing Library: http://ngslib.genome.tugraz.at
http://seqanswers.com
http://www.broadinstitute.org/gsa/wiki/index.php/File:Ngs_tutorial_depristo_1210.pdf
ANALYSIS PIPELINE
- Convert Illumina Fastq >> Sanger Fastq
- QC raw Data
- Mapping(BWA, QC-BWA, Novoalign)
- SAM To BAM
- Local realignment around Indels
- Remove Duplicates
- Base Quality Score Recalibration
- Analysis-ready Indels & SNPs
ANALYSIS PIPELINE
- Convert Illumina Fastq >> Sanger Fastq
maq ill2sanger s_1_1_sequence.txt foo.1.sanger.fastq
maq ill2sanger s_1_2_sequence.txt foo.2.sanger.fastq
FastQC: Provides a simple way to do some quality control checks on raw sequence data.
Quick impression of whether the data has problems.
Import! of data from BAM, SAM or FastQ
Summary graphs and tables to quickly assess your data
Export of results to an HTML based permanent report
Offline operation to allow automated generation of reports without running the interactive application
fastqc foo.1.sanger.fastq;
fastqc foo.2.sanger.fastq;
fastqc foo.2.sanger.fastq;
http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/
- Mapping(BWA, QC-BWA, Novoalign)
## Align with BWA
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: bwa aln -t 8 $REF foo.1.sanger.fastq > foo.1.sai;
$: bwa aln -t 8 $REF foo.2.sanger.fastq > foo.2.sai;
## Generate alignment in the SAM format
$: bwa sampe $REF foo.1.sai foo.2.sai foo.1.sanger.fastq foo.2.sanger.fastq > foo.bwa.raw.sam;
## Sort bwa SAM file using PICARD TOOLS SortSam.jar - this will also produce the BAM file
$: java -jar SortSam.jar SORT_ORDER=coordinate VALIDATION_STRINGENCY=SILENT \
INPUT= foo.bwa.raw.sam OUTPUT= foo.bwa.raw.bam;
## samtools index
$: samtools index foo.novo.raw.bam;
Use option -q15 if the quality is poor at the 3' end of reads
http://bio-bwa.sourceforge.net/bwa.shtml
BWA: with pre-alignment QC.1
BWA: with pre-alignment QC.1
Fastx: http://hannonlab.cshl.edu/fastx_toolkit
QC filter raw sequence data
always use -Q 33 for sanger phred scaled data (-Q 64)
$: cat foo.1.sanger.fastq | \
fastx_clipper -Q 33 -l 20 -v -a ACACTCTTTCCCTACACGACGCTCTTCCGATCT | \
fastx_clipper -Q 33 -l 20 -v -a CGGTCTCGGCATTCCTACTGAACCGCTCTTCCGATCT | \
fastx_clipper -Q 33 -l 20 -v -a ATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC | \
fastx_clipper -Q 33 -l 20 -v –a CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATC | \
fastq_quality_trimmer -Q 33 -t 20 -l 20 -v | \
fastx_artifacts_filter -Q 33 -v | \
fastq_quality_filter -Q 33 -q 20 -p 50 -v -o foo.1.sanger.qc.fastq;
$: cat foo.2.sanger.fastq | \
fastx_clipper -Q 33 -l 20 -v -a ACACTCTTTCCCTACACGACGCTCTTCCGATCT | \
fastx_clipper -Q 33 -l 20 -v -a CGGTCTCGGCATTCCTACTGAACCGCTCTTCCGATCT | \
fastx_clipper -Q 33 -l 20 –v -a ATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC | \
fastx_clipper -Q 33 -l 20 -v –a CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATC | \
fastq_quality_trimmer -Q 33 -t 20 -l 20 -v | \
fastx_artifacts_filter -Q 33 -v | \
fastq_quality_filter -Q 33 -q 20 -p 50 -v -o foo.2.sanger.qc.fastq;#
BWA : with pre-alignment QC.2
Compare QCd fastq files
One end of each read could be filtered out in QC
BWA cant deal with mixed PE & SE data
Need to id reads that are still paired after QC
Need to id reads that are no longer paired after QC
$: perl cmpfastq.pl foo.1.sanger.qc.fastq foo.2.sanger.qc.fastq
Reads matched on presence/absence of id's in each file :
foo.1.sanger.qc.fastq : @315ARAAXX090414:8:1:567:552#1
foo.2.sanger.qc.fastq : @315ARAAXX090414:8:1:567:552#2
Output: 2 files for each *QC.fastq file
foo.1.sanger.qc.fastq-common-out (reads in foo.1.* == reads in foo.2.*)
foo.1.sanger.qc.fastq-unique-out (reads in foo.1.* not in foo.2.*)
foo.2.sanger.qc.fastq-commont-out
foo.2.sanger.qc.fastq-unique-out
http://compbio.brc.iop.kcl.ac.uk/software/cmpfastq.php
BWA : with pre-alignment QC.3
Align with BWA
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: bwa aln -t 8 $REF foo.1.sanger.qc.fastq-common-out > foo.1.common.sai;
$: bwa aln -t 8 $REF foo.2.sanger.qc.fastq-common-out > foo.2.common.sai;
$: bwa aln -t 8 $REF foo.1.sanger.qc.fastq-unique-out > foo.1.unique.sai;
$: bwa aln -t 8 $REF foo.1.sanger.qc.fastq-unique-ou > foo.2.unique.sai;
Multi threading option : -t N
http://bio-bwa.sourceforge.net/bwa.shtml
BWA : with pre-alignment QC.4
Generate alignments in the SAM/BAM format
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
## bwa sampe for *common*
$: bwa sampe $REF foo.1.common.sai foo.2.common.sai foo.1.sanger.qc.fastq-common-out foo.2.sanger.qc.fastq-common-out > foo.common.sam;
## bwa samse for *unique*
$: bwa samse $REF foo.1.unique.sai foo.1.sanger.qc.fastq-unique-out > foo.1.unique.sam;
$: bwa samse $REF foo.2.unique.sai foo.2.sanger.qc.fastq-unique-out > foo.2.unique.sam;
## merge SAM files using PICARD TOOLS MergeSamFiles.jar - this will also sort the BAM file
$: java -jar MergeSamFiles.jar INPUT=foo.common.sam INPUT=foo.1.unique.sam INPUT=foo.2.unique.sam ASSUME_SORTED=false VALIDATION_STRINGENCY=SILENT OUTPUT=foo.bwa.raw.bam;
## samtools index
samtools index foo.bwa.raw.bam;
Details SAM/BAM Format : http://samtools.sourceforge.net/SAM1.pdf
Post Aligment processing of BAM files
Local realignment around Indels
Remove duplicate reads
Base Quality Score Recalibration
GATK: http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
PICARD TOOLS: http://picard.sourceforge.net
SAMTOOLS: http://samtools.sourceforge.net
Many other quality stats/options for processing files using these tools : see web documentation
- SAM To BAM
Sequence aligners are unable to perfectly map reads containing insertions or deletions
Alignment artefacts
False positives SNPs
Steps to the realignment process:
Step 1: Determining (small) suspicious intervals which are likely in need of realignment
Step 2: Running the realigner over those intervals
Step 3: Fixing the mate pairs of realigned reads
http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels
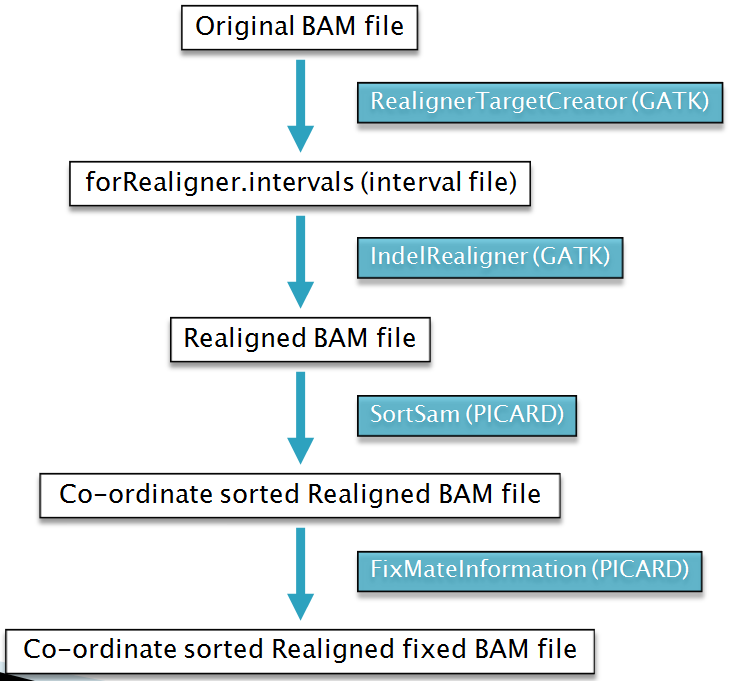
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
$: TMPDIR=~/scratch/tmp
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
## 1. Creating Intervals : RealignerTargetCreator
$: java –Xmx5g -jar $GATK -T RealignerTargetCreator -R $REF -D $ROD \
-I foo.novo.bam -o foo.novo.bam.forRealigner.intervals;
## 2. Realigning : IndelRealigner
$: java -Djava.io.tmpdir=$TMPDIR –Xmx5g -jar $GATK -T IndelRealigner \
-R $REF -D $ROD -I foo.novo.bam -o foo.novo.realn.bam \
-targetIntervals foo.novo.bam.forRealigner.intervals;
## samtools index
$: samtools index foo.novo.realn.bam;
## 3. Sort realigned BAM file using PICARD TOOLS SortSam.jar
## GATK IndelRealigner produces a name sorted BAM
$: java –Xmx5g -jar SortSam.jar \
INPUT= foo.novo.realn.bam OUTPUT=foo.novo.realn.sorted.bam \
SORT_ORDER=coordinate TMP_DIR=$TMPDIR VALIDATION_STRINGENCY=SILENT;
## samtools index
$: samtools index foo.novo.realn.soretd.bam;
## 4. Fixing the mate pairs of realigned reads using Picard tools FixMateInformation.jar
$: java -Djava.io.tmpdir=$TMPDIR -jar -Xmx6g FixMateInformation.jar \
INPUT= foo.novo.realn.sorted.bam OUTPUT= foo.novo.realn.sorted.fixed.bam \
SO=coordinate VALIDATION_STRINGENCY=SILENT TMP_DIR=$TMPDIR;
## samtools index
samtools index foo.novo.realn.sorted.fixed.bam ;
Examine aligned records in the supplied SAM or BAM file to locate duplicate molecules and remove them
$: TMPDIR=~/scratch/tmp
## Remove duplicate reads with Picard tools MarkDuplicates.jar
$: java -Xmx6g –jar MarkDuplicates.jar \
INPUT= foo.novo.realn.sorted.fixed.bam \
OUTPUT= foo.novo.realn.duperemoved.bam \
METRICS_FILE=foo.novo.realn.Duplicate.metrics.file \
REMOVE_DUPLICATES=true \
ASSUME_SORTED=false TMP_DIR=$TMPDIR \
VALIDATION_STRINGENCY=SILENT;
## samtools index
samtools index foo.novo.realn.duperemoved.bam;
- Base Quality Score Recalibration
Base Quality Score Recalibration.1
Correct for variation in quality with machine cycle and sequence context
Correct for variation in quality with machine cycle and sequence context
Recalibrated quality scores are more accurate
Closer to the actual probability of mismatching the reference genome
Done by analysing the covariation among several features of a base
Reported quality score
The position within the read
The preceding and current nucleotide (sequencing chemistry effect) observed by the sequencing machine
Probability of mismatching the reference genome
Known SNPs taken into account (dbSNP 131)
Covariates are then used to recalibrate the quality scores of all reads in a BAM file
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration
Base Quality Score Recalibration.2
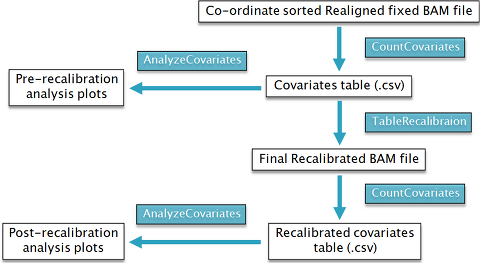
Base Quality Score Recalibration.3
Base Quality Score Recalibration.2
Base Quality Score Recalibration.3
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
## 1. GATK CountCovariates
java -Xmx8g -jar $GATK -T CountCovariates -R $REF --DBSNP $ROD \
-I foo.novo.realn.duperemoved.bam \
-recalFile foo.novo.realn.duperemoved.bam.recal_data.csv \
--default_platform Illumina \
-cov ReadGroupCovariate \
-cov QualityScoreCovariate \
-cov CycleCovariate \
-cov DinucCovariate \
-cov TileCovariate \
-cov HomopolymerCovariate \
-nback 5;
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration
Base Quality Score Recalibration.4
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: GATKacov=/share/apps/gatk_20100930/Sting/dist/AnalyzeCovariates.jar
$: GATKR=/share/apps/gatk_20100930/Sting/R
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
$: Rbin=/share/apps/R_current/bin/Rscript
## 2. GATK AnalyzeCovariates
java -Xmx5g –jar $GATKacov \
-recalFile foo.novo.realn.duperemoved.bam.recal_data.csv \
-outputDir foo.novo.realn.duperemoved.bam.recal.plots \
-resources $GATKR \
-Rscript $Rbin;
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration
Base Quality Score Recalibration.5
Base Quality Score Recalibration.5
Generate the final analysis ready BAM file for Variant Discovery and Genotyping
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
## 3. GATK TableRecalibration
$: java –Xmx6g -jar $GATK -T TableRecalibration -R $REF \
-I foo.novo.realn.duperemoved.bam \
--out foo.novo.final.bam \
-recalFile foo.novo.realn.duperemoved.bam.recal_data.csv \
--default_platform Illumina;
##samtools index
$: samtools index foo.novo.final.bam;
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration
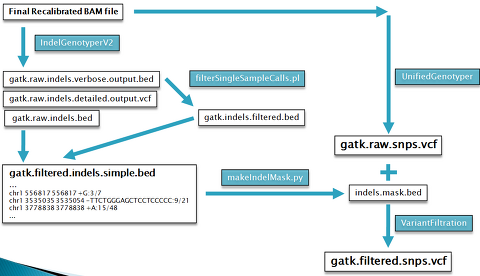
- Analysis-ready Indels & SNPs
hort Indels (GATK IndelGenotyperV2)
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: GATKPERL=/share/apps/gatk_20100930/Sting/perl
$: GATKPYTHON=/share/apps/gatk_20100930/Sting/python
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
## Generate raw indel calls
$: java -Xmx6g -jar $GATK -T IndelGenotyperV2 -R $REF --DBSNP $ROD \
-I foo.novo.final.bam \
-bed foo.gatk.raw.indels.bed \
-o foo.gatk.raw.indels.detailed.output.vcf \
--metrics_file foo.gatk.raw.indels.metrics.file \
-verbose foo.gatk.raw.indels.verbose.output.bed \
-minCoverage 8 -S SILENT –mnr 1000000;
## Filter raw indels
$: perl $GATKPERL/filterSingleSampleCalls.pl --calls foo.gatk.raw.indels.verbose.output.bed \
--max_cons_av_mm 3.0 --max_cons_nqs_av_mm 0.5 --mode ANNOTATE > foo.gatk.filtered.indels.bed
http://www.broadinstitute.org/gsa/wiki/index.php/Indel_Genotyper_V2.0
Creating an indel mask file
The output of the IndelGenotyper is used to mask out SNPs near indels.
“makeIndelMask.py” creates a bed file representing the masking intervals based on the output of IndelGenotyper.
$: GATKPYTHON=/share/apps/gatk_20100930/Sting/python
## Create indel mask file
$: python $GATKPYTHON/makeIndelMask.py foo.gatk.raw.indels.bed 5 indels.mask.bed
The number in this command stands for the number of bases that will be included on either side of the indel.
http://www.broadinstitute.org/gsa/wiki/index.php/Indel_Genotyper_V2.0
SNP Calling (GATK UnifiedGenotyper)
SNP Calling (GATK UnifiedGenotyper)
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
## SNP Calling
$: java -Xmx5g -jar $GATK -T UnifiedGenotyper -R $REF -D $ROD \
-baq CALCULATE_AS_NECESSARY -baqGOP 30 -nt 8 \
-A DepthOfCoverage -A AlleleBalance -A HaplotypeScore -A HomopolymerRun -A MappingQualityZero -A QualByDepth -A RMSMappingQuality -A SpanningDeletions \
-I foo.novo.final.bam -o foo.gatk.raw.snps.vcf \
-verbose foo.gatk.raw.snps.vcf.verbose -metrics foo.gatk.raw.snps.vcf.metrics;
This results in a VCF (variant call format) file containing raw SNPs.
VCF is a text file format. It contains meta-information lines, a header line, and then data lines each containing information about a position in the genome (SNP/INDEL calls).
http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-40
http://www.broadinstitute.org/gsa/wiki/index.php/Unified_genotyper
SNP Filtering & annotation (GATK VariantFiltration)
VariantFiltration is used annotate suspicious calls from VCF files based on their failing given filters.
It annotates the FILTER field of VCF files for records that fail any one of several filters:
Variants that lie in clusters, using the specified values to define a cluster (i.e. the number of variants and the window size).
Any variant which overlaps entries from a masking rod.
Any variant whose INFO field annotations match a specified expression! (i.e. the expression! is used to describe records which should be filtered out).
## set env variables
$: GATK=/share/apps/gatk_20100930/Sting/dist/GenomeAnalysisTK.jar
$: REF=/scratch/data/reference_genomes/human/human_g1k_v37.fasta
$: ROD=/scratch/data/reference_genomes/human/dbsnp_131_b37.final.rod
## VariantFiltration & annotation
$: java –Xmx5g -jar $GATK -T VariantFiltration -R $REF -D $ROD \
-o foo.gatk.VariantFiltration.snps.vcf \
-B variant,VCF, foo.gatk.raw.snps.vcf \
-B mask,Bed, indels.mask.bed --maskName InDel \
--clusterSize 3 --clusterWindowSize 10 \
--filterExpression! "DP <= 8" --filterName "DP8" \
--filterExpression! "SB > -0.10" --filterName "StrandBias" \
--filterExpression! "HRun > 8" --filterName "HRun8" \
--filterExpression! "QD < 5.0" --filterName "QD5" \
--filterExpression! "MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)" --filterName "hard2validate";
More information on the parameters used can be found in: http://www.broadinstitute.org/gsa/wiki/index.php/VariantFiltrationWalker http://www.broadinstitute.org/gsa/wiki/index.php/Using_JEXL_expressions
SNP Filtering: VCFTOOLS
VCFTOOLS: methods for working with VCF files: filtering,validating, merging, comparing and calculate some basic population genetic statistics.
Example of some basic hard filtering:
## filter poor quality & suspicious SNP calls
vcftools --vcf foo.gatk.VariantFiltration.snps.vcf \
--remove-filtered DP8 --remove-filtered StrandBias --remove-filtered LowQual \
--remove-filtered hard2validate --remove-filtered SnpCluster \
--keep-INFO AC --keep-INFO AF --keep-INFO AN --keep-INFO DB \
--keep-INFO DP --keep-INFO DS --keep-INFO Dels --keep-INFO HRun --keep-INFO HaplotypeScore --keep-INFO MQ --keep-INFO MQ0 --keep-INFO QD --keep-INFO SB --out foo.gatk.good.snps ;
this produces the file " foo.gatk.good.snps.recode.vcf"
VCFTOOLS for SNP QC and statistics
VCFTOOLS can be used to generate useful QC measures, PLINK ped/map, Impute input and more....
## QC & info
$: for MYQC in missing freq2 counts2 depth site-depth site-mean-depth geno-depth het hardy singletons;do
vcftools --vcf foo.gatk.good.snps.recode.vcf --$MYQC \
--out foo.gatk.good.snps.QC;
done
## write genotypes, genotype qualities and genotype depth to separate files
$: for MYFORMAT in GT GQ DP;do
vcftools --vcf foo.gatk.good.snps.recode.vcf \
--extract-FORMAT-info $MYFORMAT --out foo.gatk.good.snps;
done
## make PLINK ped and map files
vcftools --vcf foo.gatk.good.snps.recode.vcf --plink --out foo.gatk.good.snps
http://vcftools.sourceforge.net/
More useful links:
More useful links:
http://www.broadinstitute.org/gsa/wiki/index.php/Prerequisites
http://www.broadinstitute.org/gsa/wiki/index.php/Building_the_GATK
http://www.broadinstitute.org/gsa/wiki/index.php/Downloading_the_GATK
http://www.broadinstitute.org/gsa/wiki/index.php/Input_files_for_the_GATK
http://www.broadinstitute.org/gsa/wiki/index.php/Preparing_the_essential_GATK_input_files:_the_reference_genome
http://www.broadinstitute.org/gsa/wiki/index.php/The_DBSNP_rod
没有评论:
发表评论